Areas of research
Our group studies patterns of rare genetic variation in large collections of human genomic data, both from patients and reference population individuals, and designs tools and methods to help interpret that variation. We are focused on moving from studying single variants at a time to understanding how they impact disease in their genomic context.

Identifying constrained genomic regions
A long-standing interest in our group has been using standing variation in the general population to identify those genes, and genomic regions, that are import to human health. We have previously developed scores that measure a gene’s tolerance to being mutated, such as the pLI score. These scores have highlighted thousands of genes that appear to be ‘constrained’ against damaging variation in the general population, and have been found to be enriched for mutations in patients with severe disease.

Improving variant interpretation
A major challenge in medical genetics is interpreting the potential impacts of variants, which often requires integrating multiple lines of evidence to determine their consequences on disease risk. We aim to improve variant interpretation by using gene-level, region-level, and variant-level information when considering the potential pathogenicity of variants. These improvements are desperately needed to improve clinical diagnostic rates, which remain ~50%.
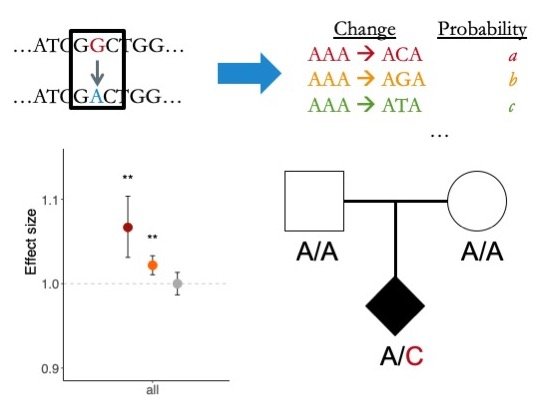
Associating rare variation with disease
One of our central goals has been to develop robust statistical frameworks to associate rare variation with risk for disease. In particular, we have created methods to identify genes that are significantly burdened by de novo (newly arising) mutations. These methods have helped identify dozens of disease-associated genes for autism spectrum disorders, congenital heart disease, and developmental disorders, among others.